目前发现的耐药机制,解释了部分遗传学和组织学耐药原因,但涉及层面有限,其他维度的机制还存在诸多未解之谜;对第三代抑制剂耐药机制的了解,则更为匮乏,即使发现的部分耐药原因,也缺乏针对性药物上市予以克服,一旦出现耐药则应对手段极为有限。揭示耐药新机制、寻找克服耐药新靶点,是肿瘤科学的前沿问题,也是研发克服耐药新型药物的前提,具有重要的科学价值和临床转化意义。
2021年10月7日,上海交通大学医学院药理学与化学生物学系朱亮课题组在 Science 子刊 Science Translational Medicine 期刊在线发表了题为:Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR- targeted therapy in lung cancer 的研究成果,该研究揭示了肺癌EGFR-TKI耐药新机制并提示克服耐药新治疗手段。

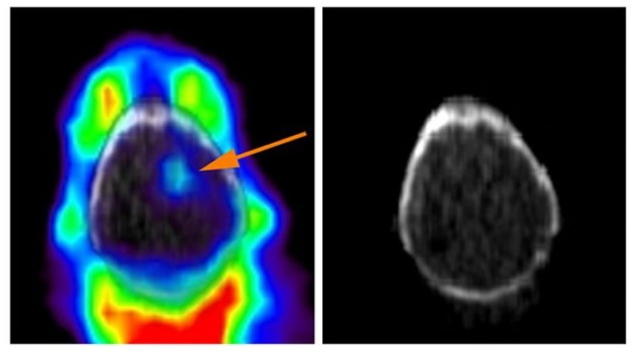
课题组基于前期研究结果,结合转录组学分析,在符合临床治疗路径的体内体外耐药模型及临床耐药复发数据库中筛选出与耐药相关的蛋白分子AKR1B1,功能性研究发现一代和三代EGFR-TKI耐药的多种肿瘤细胞模型,其存活、生长和药物抵抗均依赖于AKR1B1,反之,原先对EGFR-TKI敏感的肿瘤细胞,若高表达该分子,则在体外和体内均获得耐药能力。
基因组和代谢组学整合分析表明耐药细胞普遍发生代谢重组现象,非靶向代谢组学检测提示其中谷胱甘肽代谢途径变化最为显著,随后的靶向代谢组学分析发现谷胱甘肽从头合成代谢通路在耐药细胞更为活跃,同时,耐药复发肿瘤病人血液中、耐药复发小鼠肿瘤组织和血液中还原性和氧化型谷胱甘肽也更为富集。同位素示踪代谢流分析显示耐药细胞对环境中胱氨酸的摄取能力以及随后由胱氨酸流向谷胱甘肽从头合成代谢的能力增强。
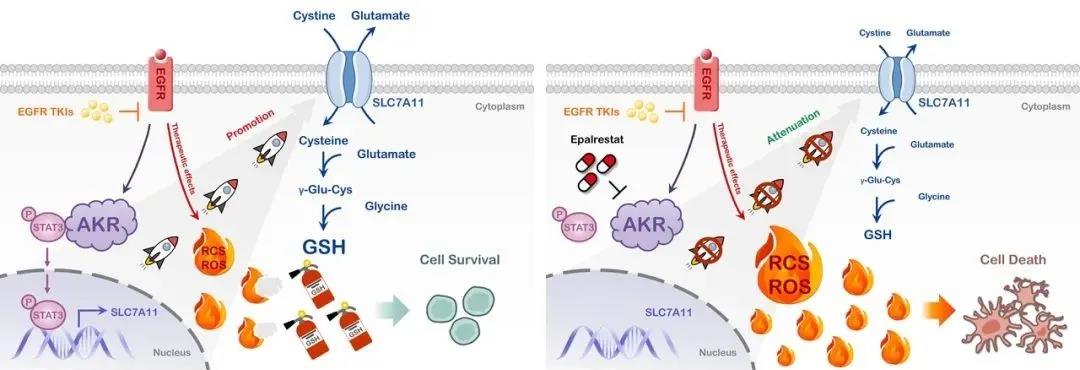
该代谢机制将耐药细胞中活性氧(ROS)和活性羰基化合物(RCS)维持在较低水平,并决定耐药细胞对EGFR-TKI的抵抗能力。以乙酰半胱氨酸(NAC)喂饲小鼠,其负荷的肿瘤对多种EGFR-TKI耐药,停止NAC喂饲后该耐药特性消退。耐药细胞增强的胱氨酸摄入和谷胱甘肽从头合成受AKR1B1调控,用遗传学或药理学手段抑制AKR1B1可取消上述代谢特征并且逆转耐药,反之若敏感细胞高表达AKR1B1则使谷胱甘肽从头合成增强并获得耐药性。
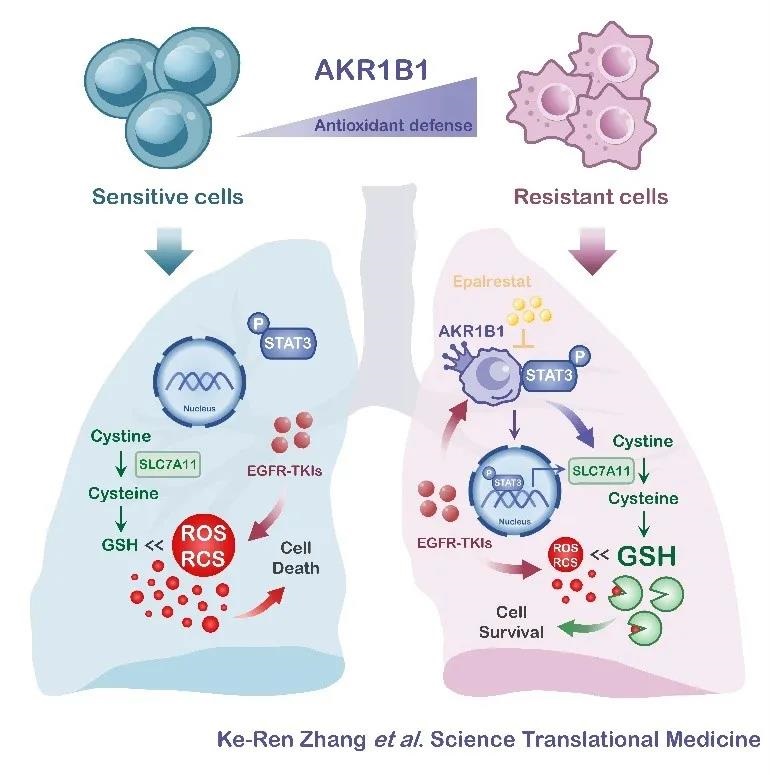
进一步研究表明TKI药物压力诱导AKR1B1上调,AKR1B1与转录因子STAT3发生相互作用而使后者激活,进入细胞核,增加靶基因SLC7A11转录。SLC7A11的蛋白产物细胞膜胱氨酸转运体上调后使细胞摄入胱氨酸增加,加强谷胱甘肽从头合成,淬灭活性氧(ROS)和活性羰基化合物(RCS),提高肿瘤细胞应对药物压力的能力,从而产生耐药(图1)。
鉴于AKR1B1对耐药的驱动和维持作用,课题组基于适应症重定位策略,应用其已上市用于治疗糖尿病神经病变的选择性抑制剂依帕司他予以干预,可使耐药细胞重新获得对一、三代EGFR-TKI的敏感性,并且在负荷人源肿瘤细胞系来源移植瘤(CDX)和人源肿瘤组织来源移植瘤(PDX)的小鼠体内阻止肿瘤耐药的产生。
图1:肺癌EGFR-TKI耐药的AKR依赖性代谢重编程机制及其逆转策略
该研究从代谢维度发现肺癌EGFR靶向治疗耐药的新机制,同时也提示实体瘤靶向药耐药存在代谢重编程驱动因素。所发现的AKR关键节点是EGFR-TKI多代靶向药耐药的共有机制,药理学干预后在临床前实验中显示预防及克服耐药作用,甚至对棘手的第三代药物奥希替尼耐药亦有效。Science Translational Medicine同期配发编者按,提示该研究的新发现及抗糖尿病药物重定位对克服肺癌EGFR靶向药耐药治疗策略的启示作用。
论文链接:
https://www.science.org/doi/10.1126/scitranslmed.abg6428